Reference sequences
Mus musculus
The references sequences are TRA@.gb, TRB@.gb and TRG@.gb, downloaded in GenBank format from RefSeq. To save the files with full information from the NCBI's website, select Show sequence and GenBank (full) as shown in the panels below.
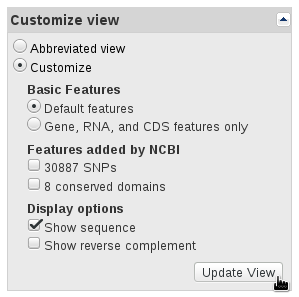

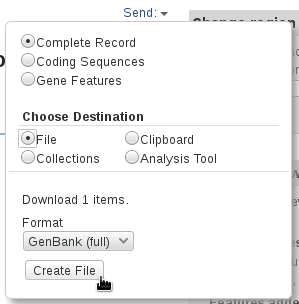

The V, (D), J and C segments were extracted with the command make
gb2fa, wich uses the the extractfeat program of the EMBOSS package.
This directory contains V and J segments aligned on conserved motifs in the files V.fa and J.fa. These alignments were made by hand using SeaView and may need to be revised when the reference sequences change.
The files V-C.fa, V after C.fa, J-FGxG.fa, J before FGxG.fa are
derived from the manually edited files V.fa and J.fa described above,
by removing everything after or before their conserved motif, followed by
degapping with make degap.
A BWA index is provided for the V segments trunkated after the conserved
cysteine, in the V directory, and can be refreshed with the command make
bwa-index. It is used by the command clonotypeR detect.
Other organisms
Work to support the analysis of repertoires from other species is under way.
The current workaround is to download reference loci from GenBank, overwrite
the files TRA@.gb, TRB@.gb and TRG@.gb, and repeat the process described
above for mouse sequences. For human, the accession numbers are
NG_001332, NG_001333, and NG_001336.
Redundant sequences
Some V segments have identical sequences. This is incompatible with clonotypeR's detection strategy, based on mapping qualities.
Mapping qualities are an
estimation of the probability that a genomic alignment is incorrect. If two
V segments are identical, a read can align to both with equal probability,
and therefore the mapping quality will be low, in the sense that it is
impossible to know precisely from which V segment the RNA was transcribed.
ClonotypeR uses mapping quality scores to distinguish between closely related
V segments, and by default discards reads where the mapping quality is too
low. Therefore, redundant references sequences were removed from the V.fa
alignment.
Removed redundant V segments are recorded in the file V.removed and were
detected with the command export SEQ_LIST=$(infoseq V.fa -filter -only -usa
-noheading) ; for SEQ1 in $SEQ_LIST ; do for SEQ2 in $SEQ_LIST ; do if ! [
$SEQ1 = $SEQ2 ]; then needle --filter $SEQ1 $SEQ2 2> /dev/null | grep -B10
'100.0' ; fi; done; done. Note that with some PCR designs, more V segments
appear identical. You may need to correct V.fa and V.removed accordingly,
or turn off the use of mapping qualities in the R command clonotype_table.